
Molecular Orbital Theory (MOT) is a sophisticated approach to understanding the electronic structure of molecules. Developed in the early 20th century by scientists like Friedrich Hund and Robert S. Mulliken, it revolutionized our perception of chemical bonding by moving beyond the localized electron pair concept of Valence Bond Theory (VBT). In MOT, electrons are not considered to be confined to individual bonds between two atoms, but rather are treated as moving under the influence of all the atomic nuclei in the entire molecule. This leads to the formation of molecular orbitals (MOs) that span the entire molecular framework, providing a more accurate and nuanced description of bonding, especially for complex systems.
Core Principles of Molecular Orbital Theory
At its heart, MOT is built upon several fundamental principles:
Formation of Molecular Orbitals from Atomic Orbitals:
When atoms combine to form a molecule, their atomic orbitals (AOs) cease to exist in their original form. Instead, they interact and combine to form new orbitals called molecular orbitals (MOs). This combination is often described by the Linear Combination of Atomic Orbitals (LCAO) method, where molecular orbitals are expressed as mathematical sums and differences of atomic orbitals.
For two atomic orbitals (ψA and ψB) to combine effectively, they must:
- Have similar energies. For example, a 1s orbital of one atom can combine with a 1s orbital of another, but a 1s and a 2p orbital generally won’t combine significantly due to their large energy difference.
- Have appropriate symmetry. The atomic orbitals must have the correct spatial orientation to overlap effectively.
- Overlap significantly in space. The greater the overlap, the stronger the interaction.
Number of Molecular Orbitals Formed:
The total number of molecular orbitals formed is always equal to the total number of atomic orbitals that combine. If n atomic orbitals combine, n molecular orbitals will be generated. For instance, when two atomic orbitals combine, they form two molecular orbitals: one bonding and one antibonding.
Types of Molecular Orbitals:
Bonding Molecular Orbitals (σ, π): These are formed by the constructive interference of atomic orbitals, meaning their wave functions add up when they are in phase. This leads to an increased electron density in the region between the nuclei, which stabilizes the molecule by increasing the attractive forces between the electrons and the nuclei. Bonding MOs are lower in energy than the original atomic orbitals.
- Sigma (σ) orbitals: Formed by head-on (axial) overlap of atomic orbitals (e.g., s-s, s-p, pz-pz). Electron density is concentrated along the internuclear axis.
- Pi (π) orbitals: Formed by lateral (sideways) overlap of parallel p orbitals (e.g., px-px, py-py). Electron density is concentrated above and below the internuclear axis, with a nodal plane along the axis.
Antibonding Molecular Orbitals (σ∗, π∗): These are formed by the destructive interference of atomic orbitals, where their wave functions subtract when they are out of phase. This results in a decreased electron density between the nuclei and an increased electron density outside the internuclear region, leading to repulsion between the nuclei and destabilization of the molecule. Antibonding MOs are higher in energy than the original atomic orbitals. They are often denoted with an asterisk (*).
Non-bonding Molecular Orbitals: In some cases, atomic orbitals may not interact effectively with other atomic orbitals due to symmetry or energy mismatch. These form non-bonding molecular orbitals, whose energy is approximately the same as the original atomic orbital, and they do not significantly contribute to or detract from the bond strength.
Filling of Molecular Orbitals:
Electrons are filled into molecular orbitals according to the same rules that govern the filling of atomic orbitals:
- Aufbau Principle: Electrons fill molecular orbitals in order of increasing energy.
- Pauli Exclusion Principle: Each molecular orbital can hold a maximum of two electrons, and these electrons must have opposite spins.
- Hund’s Rule of Maximum Multiplicity: For degenerate molecular orbitals (orbitals with the same energy), electrons will occupy each orbital individually with parallel spins before any orbital is doubly occupied.
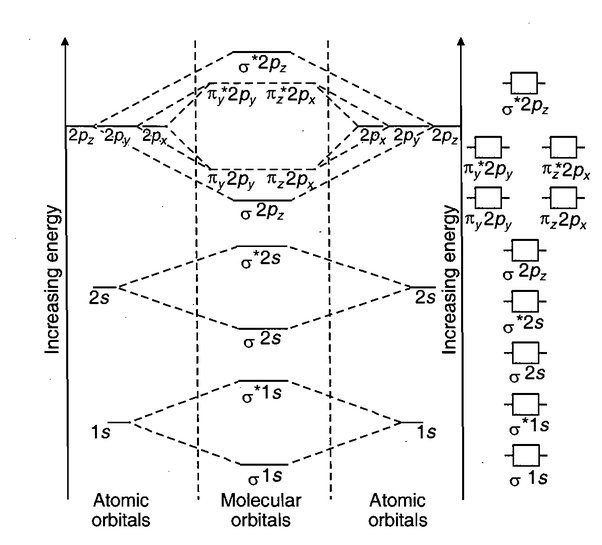
Polycentric Nature of Molecular Orbitals:
Unlike atomic orbitals, which are monocentric (associated with a single nucleus), molecular orbitals are polycentric, meaning the electrons in them are influenced by multiple nuclei in the molecule. This concept of electron delocalization is a key strength of MOT.
Applications and Advantages of Molecular Orbital Theory
MOT provides a powerful framework for explaining and predicting various molecular properties that Valence Bond Theory struggles with:
- Magnetic Properties: One of the most significant triumphs of MOT is its ability to correctly predict the magnetic properties of molecules, particularly the paramagnetic nature of dioxygen (O2). Valence Bond Theory predicts O2 to be diamagnetic (all electrons paired), but experimental evidence shows it is paramagnetic (attracted to a magnetic field, indicating unpaired electrons). MOT, through its molecular orbital diagram, clearly shows two unpaired electrons in the π∗ antibonding orbitals of O2, explaining its paramagnetism.
- Bond Order and Stability: MOT allows for the calculation of bond order, which is a measure of the net number of bonds between two atoms: Bond Order = 21 (Number of electrons in bonding MOs−Number of electrons in antibonding MOs) A higher bond order indicates a stronger and more stable bond, and a shorter bond length. For example, H2 has a bond order of 1, while He2 has a bond order of 0, correctly predicting that H2 is stable and He2 is not. This also explains why molecules like N2 (bond order 3) are exceptionally stable.
- Explaining Resonance Structures: For molecules that require multiple resonance structures in Valence Bond Theory (e.g., benzene), MOT naturally accounts for electron delocalization. In benzene, the π electrons are delocalized over the entire ring of carbon atoms, rather than being confined to alternating single and double bonds. This delocalization provides enhanced stability.
- Spectroscopic Properties: MOT is crucial for interpreting spectroscopic data, particularly in UV-Visible spectroscopy. Electronic transitions, where electrons are promoted from lower-energy molecular orbitals (like the Highest Occupied Molecular Orbital, HOMO) to higher-energy molecular orbitals (like the Lowest Unoccupied Molecular Orbital, LUMO), correspond to the absorption of light at specific wavelengths. MOT helps predict these energy gaps and thus the UV-Vis absorption spectra of molecules.
- Reactivity Prediction: The HOMO and LUMO play critical roles in understanding molecular reactivity. The HOMO represents the highest energy occupied molecular orbital, which is where electrons are most readily available for donation (nucleophilic attack). The LUMO is the lowest energy unoccupied molecular orbital, which is where electrons can be accepted (electrophilic attack). The energy gap between the HOMO and LUMO (HOMO-LUMO gap) is also indicative of a molecule’s reactivity; smaller gaps generally mean higher reactivity.
- Existence of Ions: MOT can explain the existence and stability of molecular ions like H2+, which are difficult to rationalize with Valence Bond Theory.
Comparison with Valence Bond Theory
While both MOT and VBT aim to describe chemical bonding, they approach the problem from different perspectives:
| Feature | Molecular Orbital Theory (MOT) | Valence Bond Theory (VBT) |
| Electron Delocalization | Electrons are delocalized over the entire molecule. | Electrons are localized in bonds between specific atoms. |
| Orbital Nature | Molecular orbitals are polycentric, extending over multiple nuclei. | Atomic orbitals (often hybridized) overlap to form localized bonds between two nuclei. |
| Atomic Orbital Identity | Atomic orbitals lose their individual identity upon forming MOs. | Atomic orbitals largely retain their identity, forming hybrid orbitals. |
| Resonance | Naturally explains electron delocalization; no need for multiple resonance structures. | Requires the concept of resonance (multiple Lewis structures) to describe delocalization. |
| Magnetic Properties | Accurately predicts paramagnetism (e.g., O2). | Often fails to predict correct magnetic properties (e.g., O2 predicted diamagnetic). |
| Computational Complexity | Generally more computationally intensive for larger molecules, but simpler for certain aspects. | Can be more computationally complex for systems requiring extensive resonance. |
| Focus | Describes the electronic structure of the entire molecule. | Focuses on individual bonds and their formation. |
Despite their differences, it’s important to note that at their theoretical limits, both MOT and VBT can provide equivalent descriptions of molecular structure. Each theory has its strengths and weaknesses, and chemists often use both to gain a comprehensive understanding of chemical bonding. VBT provides a more intuitive picture of localized bonds and molecular geometry (via hybridization), while MOT offers a more accurate quantum mechanical description, especially for delocalized systems and magnetic properties.
Limitations of Molecular Orbital Theory
While powerful, MOT also has its limitations:
- Complexity for Larger Molecules: For very large and complex molecules, constructing and interpreting detailed molecular orbital diagrams can become exceedingly challenging and computationally demanding.
- Approximations: The LCAO method is an approximation. More rigorous quantum mechanical calculations are often necessary for highly accurate results.
- Visualizing Orbitals: While MOs are mathematical functions, visualizing their shapes and understanding their intricate interactions can be less intuitive than the localized bonds of VBT for introductory learners.
Advanced Concepts in Molecular Orbital Theory
Beyond the basic framework, MOT extends to cover more advanced concepts:
- Hybridization (within MO context): While traditionally a VBT concept, the idea of mixing atomic orbitals to form new orbitals that are more suitable for bonding can also be incorporated into more advanced MO treatments, though it’s not a fundamental starting point as it is in VBT.
- Frontier Molecular Orbitals (HOMO and LUMO): As mentioned, these orbitals are central to understanding chemical reactions and spectroscopic transitions. The interaction between the HOMO of one molecule and the LUMO of another is a key aspect of molecular reactivity in concepts like frontier molecular orbital theory.
- Symmetry and Group Theory: For complex molecules, group theory is employed to classify molecular orbitals based on their symmetry properties, simplifying the construction of MO diagrams and understanding selection rules for spectroscopic transitions.
- Computational Chemistry: MOT forms the basis for many computational chemistry methods (e.g., Hartree-Fock, Density Functional Theory) that are used to predict molecular structures, energies, and properties with high accuracy.
In conclusion, Molecular Orbital Theory stands as a fundamental and indispensable theory in chemistry. By treating electrons as delocalized entities spanning the entire molecule, it provides a quantum mechanically sound and highly predictive framework for understanding the nature of chemical bonds, explaining phenomena like paramagnetism, resonance, and spectroscopic behavior, and serving as a bedrock for modern computational chemistry. While it presents a more abstract view of bonding than its predecessor, its explanatory power and accuracy make it an essential tool for chemists across various disciplines.



{kind=link}